

Parole d'expert
L’IDMP, Identification of Medicinal Products, est un ensemble de cinq normes ISO fournissant des éléments de données et de structures...

L’IDMP, Identification of Medicinal Products, est un ensemble de cinq normes ISO fournissant des éléments de données et de structures pour identifier et échanger de manière unique des informations sur :
La réglementation IDMP couvre l’ensemble du cycle de vie du médicament, y compris les produits en développement, les produis expérimentaux, les produits en cours d’évaluation et les produits déjà autorisés.
L’objectif de la réglementation IDMP est de simplifier l’échange d’informations entre les parties prenantes et d’améliorer l’interopérabilité des systèmes dans le réseau européen de réglementation des médicaments et au niveau international.
Bien que cette réglementation ne devienne obligatoire qu’à l’échelle européenne, elle intéresse fortement l’agence américaine (FDA), ainsi que les agences Suisse et du Japon qui vont très certainement s’inspirer des principes de l’IDMP pour leurs agences respectives.
La mise en œuvre de cette nouvelle réglementation est difficile à comprendre et à appréhender par les professionnels de l’industrie pharmaceutique. Charlélie, Responsable Affaires Réglementaires et Vigilances, revient dans cette interview sur la réglementation IDMP : De quoi s’agit-il ? Quelles sont les données qui devront être soumises électroniquement ? Il donne également ses conseils et pistes de réflexion pour réussir la mise en conformité à cette réglementation.
L’IDMP a pour but de faciliter l’identification et la traçabilité d’un produit pharmaceutique depuis son développement, sa fabrication jusqu’à l’obtention et le maintien de son Autorisation de Mise sur le Marché (AMM) en incluant le suivi de la pharmacovigilance.
A l’heure actuelle, cette identification est possible mais peut s’avérer laborieuse à cause de la localisation des données parfois dans des sources structurées comme des systèmes informatisés, ou alors dans des sources non-structurées tels que des documents électroniques ou papiers.
L’objectif de la réglementation IDMP est donc de centraliser la donnée avec une source unique, afin qu’elle soit facilement consultable et utilisable, toujours dans l’intérêt du patient.
Aujourd’hui, il est primordial de garantir une information fiable dans le secteur de la santé. C’est une industrie qui s’est digitalisée récemment. Cette évolution rapide peut engendrer une désorganisation de la donnée avec sa répétition dans différentes applications au sein d’une même société. Cela met donc en évidence un besoin important d’harmoniser, de rationaliser et de fiabiliser la donnée disponible à un temps T.
Il est important que la donnée soit harmonisée autant au sein des laboratoires que pour les organismes de santé extérieurs qui souhaitent disposer de cette information.
Le premier maillon à consolider est le référentiel de données externe aux laboratoires. C’est pourquoi, l’Europe a mis en place un référentiel de données unique (SPOR) avec des listes de vocabulaire contrôlé obligatoires dans le cadre de l’IDMP et accessibles pour l’ensemble des acteurs de l’industrie pharmaceutique.
L’objectif est de pouvoir identifier le produit sur tout son cycle de vie : où il en est, de quel produit on parle, de quoi il est composé, qui l’a fabriqué… dans le but d’avoir une traçabilité complète.
Afin d’assurer cette traçabilité complète, il y aura notamment le suivi de l’ensemble des effets indésirables (pharmacovigilance) afin de prendre des décisions rapidement mais aussi un suivi pour lutter contre la contrefaçon des médicaments. Pour cela, des identifiants de produits générés via l’IDMP seront partagés avec le système en charge de la sérialisation dans le but d’authentifier un médicament depuis sa distribution jusqu’à sa dispensation au patient.
La réglementation rentrera en vigueur au deuxième trimestre 2023 et sera obligatoire pour les produits européens qui sont enregistrés en procédure centralisée à l’European Medicines Agency (EMA).
Si le laboratoire n’est pas en conformité avec l’IDMP cela engendrera une invisibilité du produit sur le portail de l’IDMP et donc certaines obligations réglementaires ne pourront pas être réalisées. Il y aura donc un impact pour le laboratoire car le produit ne complètera pas toutes les étapes réglementaires pour obtenir son AMM et être commercialisé.
La réglementation IDMP va reposer sur 5 normes ISO.
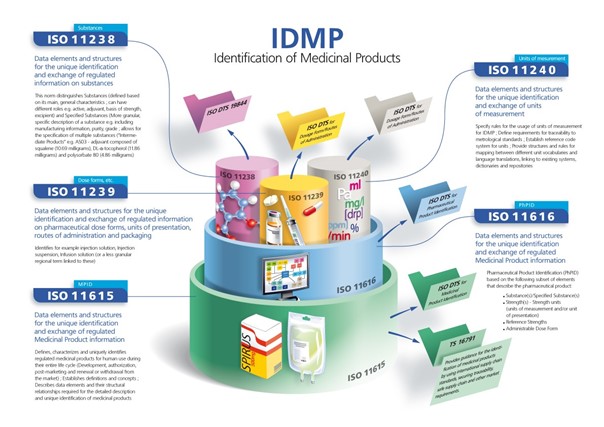
Cette norme est liée à la composition du produit c’est-à-dire quels sont ses principes actifs, ses excipients, ses adjuvants etc.
Cette norme est liée aux données qui relatent sous quelle forme le produit est présenté (comprimé, sirop, etc.), par quelle voie est-il administré et dans quels conditionnements primaires et secondaires se trouve-t-il (blister, bouteille etc.). Cette norme est relativement compliquée à mettre en place étant donné sa granularité très fine.
Cette norme peut paraître simple et pourtant, tous les pays n’utilisent pas le système métrique. Et les pays partageant les mêmes unités de mesures ont la possibilité d’exprimer différemment le dosage d’un même produit dans leur langue nationale. L’objectif étant que l’intégralité des données de mesure soient harmonisées.
L’objectif de cette norme est de centraliser et de structurer toutes les données liées au produit pharmaceutique décrit avec les normes citées ci-dessus.
Cette dernière norme permet d’avoir une vision d’ensemble du médicament en rassemblant notamment tous les identifiants générés au cours de son cycle de vie et donc de partager ses informations avec toutes les parties prenantes.
L’IDMP va mettre en place des nouveautés en termes de soumission de données. Elle va s’appuyer sur la réglementation actuelle en vigueur dont elle fait partie et augmenter le scope de données en ajoutant de nouvelles thématiques :
Aujourd’hui dans l’EVMPD, il est obligatoire de soumettre uniquement les données liées à l’indication thérapeutique, c’est-à-dire, ce pourquoi le laboratoire a obtenu son Autorisation de Mise sur le Marché (AMM), la raison pour laquelle un médecin prescrit ce médicament à ses patients.
Les normes IDMP viennent rajouter de nouvelles informations obligatoires qui devront être soumises aux autorités comme : les contre-indications, les interactions médicamenteuses et les effets indésirables (ce qui se trouve dans la notice). Toutes ces informations devront être déclarées dès la mise sur le marché du produit pharmaceutique.
Avec l’EVMPD, on ne soumet aucune information sur la fabrication d’un produit. Les normes IDMP viennent donc ajouter ces informations aux données réglementaires obligatoires. Il faudra ainsi soumettre : qui fabrique, quoi, depuis quand, et à quel moment, quelles sont ses autorisations. Et ceci à tous les niveaux : qui fabrique le produit fini, qui fabrique chaque substance (principes actifs, adjuvants…), qui fabrique la boîte en carton etc.
Il sera dorénavant obligatoire de déclarer la composition du produit dans son intégralité : le ou les principes actifs bien sûr, les excipients/adjuvants, leurs caractéristiques chimiques et structurelles. Si le produit est transformé ou non. Il existe des produits qui sont sous forme de poudre et de solvant avant leur administration.
Dans l’EVMPD, le statut de commercialisation n’est pas déclaré. Avec l’IDMP cela deviendra obligatoire, c’est-à-dire savoir à l’instant T à travers le monde où est commercialisé ou non le produit, depuis quand, s’il a été suspendu et pour quelles raisons.
A l’heure actuelle, la délivrance du médicament n’est pas déclarée dans l’EVMPD. Lors de l’entrée en vigueur des normes IDMP, il deviendra obligatoire de déclarer les données suivantes : le médicament est-il délivré avec ou sans ordonnance ? Est-il remboursé par la sécurité sociale ? Il sera alors plus facile de détecter les fraudes et les abus.
L’enjeu majeur de la mise en conformité avec cette nouvelle réglementation est de réussir à consolider la donnée. Dans les laboratoires, les données sont gérées par différents services. Très souvent chaque service à son propre système informatisé et ils ne sont pas forcément connectés entre eux. Il est donc très difficile de consolider la donnée dans un même Système d’Information (SI).
Même pour les SI qui sont connectés, on constate fréquemment qu’ils n’utilisent pas le même référentiel de données et que donc la consolidation s’avère très compliquée.
Beaucoup de données se trouvent sur papier ou dans des documents stockés dans des GED (Gestion Electronique de Document) et donc ne sont pas facilement exploitables. On appelle ça les données non-structurées car la donnée est présente mais l’extraire et l’utiliser va dépendre d’une action humaine (bien que les technologies de « text-mining » deviennent de plus en plus performantes).
Ainsi, avant toute chose, la première étape de la mise en conformité IDMP est de cartographier les SI de la société afin de savoir qui est le propriétaire de la donnée et où elle est stockée.
Souvent après cette étape on se rend compte que plusieurs services exploitent la même donnée mais de manière différente et en utilisant une liste de vocabulaire contrôlée différente. On parle de la même chose, mais en l’exprimant différemment.
Il faut donc partir de zéro et tout remettre à plat avec le soutien de la Direction comme moteur de cette transformation.
La problématique sous-jacente à la norme IDMP se résume, à la base, à une question de données de référence. Agence européenne des médicaments
La meilleure solution pour réussir sa mise en conformité IDMP est de mettre en place une gouvernance de la donnée. Celle-ci va permettre d’implémenter un système de Master Data Management (MDM), référentiel de données unique au sein d’une même société.
Grâce à un système de MDM central, il va être possible d’interfacer tous les SI et donc d’avoir une source de données fiable qui sera transmise dans le cadre de l’IDMP. L’intérêt de l’implémentation d’un MDM est également de pouvoir rationnaliser ses processus internes, améliorer la qualité des données, les échanges des données et de ce fait d’accélérer la mise sur le marché du produit grâce à une donnée fiable. Le produit sera plus vite commercialisé et le patient aura plus rapidement accès à son traitement.
Un RIMS (Regulatory Information Management System) peut tout à fait être choisi pour endosser le rôle de MDM. Outre ses fonctions principales de gestion et de suivi d’un portefeuille de produit, les éditeurs permettent de réaliser directement les soumissions IDMP. Les données concernées par les normes IDMP pourront donc être automatiquement transmises à l’EMA via ce système.
La mise en place d’un RIMS facilite donc la mise en conformité réglementaire et participe à la rationalisation des processus.
La première étape la plus cruciale pour être préparé à l'IDMP, consiste à comprendre et déterminer la localisation des données au sein de l'organisation. Il est important d’établir une stratégie globale d’entreprise pour définir une gouvernance des données et identifier les systèmes informatisés sources.
Être accompagné dans cette mise en œuvre est indispensable pour réussir sa mise en conformité IDMP. Chez Infogene, nous sommes une ESN Santé regroupant les compétences IT, métier, réglementaires et qualité nécessaires à la réussite de votre projet. Nous intervenons sur tout le cycle de vie du projet grâce à nos experts en data management, affaires réglementaires, qualité et validation des systèmes GxP de notre filiale Pharmasys. Vous trouverez en un seul acteur l’ensemble des compétences requises à la réussite de votre mise en conformité. Nous avons de multiples références dans ce domaine et comprenons vos enjeux et besoins.
N’hésitez pas à nous contacter afin que nous puissions échanger sur votre projet et vous présenter nos références du secteur.


